AI Identifying Unknown Genetic Mutations Causing Rare Diseases
Introduction
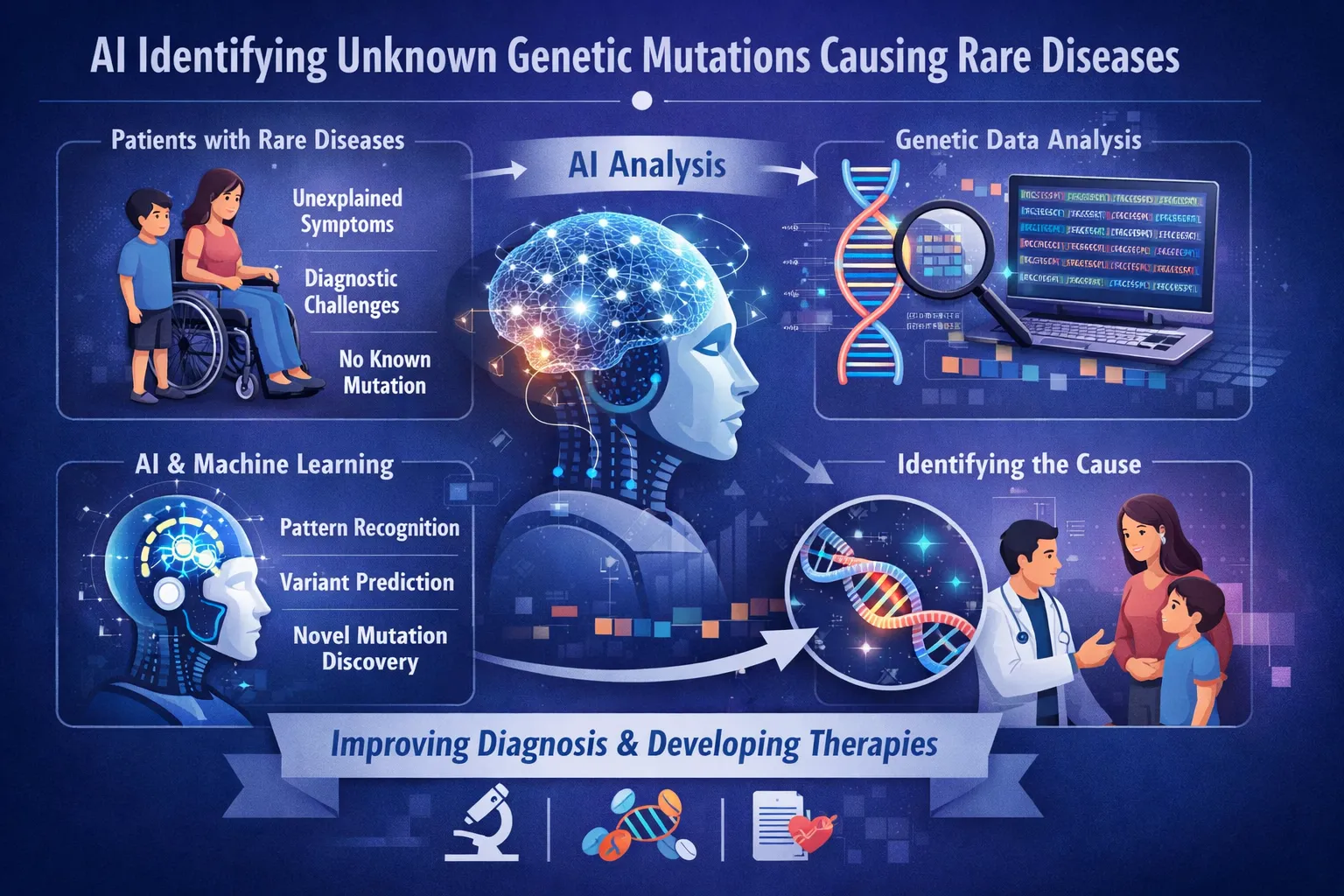
The human genome contains roughly 20,000 genes, yet for approximately half of all individuals with a rare disease, no clear genetic cause is ever identified. The challenge of identifying unknown genetic mutations that cause rare diseases has long frustrated clinicians and researchers. Each human genome contains tens of thousands of genetic variants, yet only handfuls typically drive disease pathology. Traditional methods struggle to distinguish harmful mutations from benign ones, particularly when those mutations have never been documented in any patient before. This diagnostic gap is precisely where artificial intelligence is now demonstrating measurable impact. AI identifying unknown genetic mutations causing rare diseases represents a fundamental shift from pattern-matching against known cases to predicting pathogenicity from first principles. The field has moved rapidly from theoretical possibility to clinical application, with multiple validated tools now in real-world use. AI identifying unknown genetic mutations causing rare diseases directly addresses this limitation by predicting pathogenicity from biological principles rather than historical records.For the 350 million people worldwide affected by rare diseases, AI identifying unknown genetic mutations causing rare diseases represents the most promising pathway to diagnosis, treatment, and ultimately better outcomes.

AI Identifying Unknown Genetic Mutations Causing Rare Diseases
How Evolutionary AI Models Detect Harmful Mutations Without Prior Human Cases
Evolution has been conducting mutation experiments for billions of years. Every living species carries a record of which protein changes are tolerated and which are lethal. Modern AI models leverage this evolutionary data to assess human genetic variants, even those never seen before in any patient.
The popEVE model, developed by researchers at Harvard Medical School and the Centre for Genomic Regulation, exemplifies this approach. It compares protein sequences across hundreds of thousands of species to identify which amino acid positions are essential for life. When a human mutation appears at a position consistently preserved across evolution, the model flags it as likely harmful. When the position shows natural variation across species, the mutation is more likely benign.
This evolutionary framework solves a critical problem. For ultra-rare conditions, there are no case histories to consult. Even with global genome sequencing efforts, some patients present mutations that are genuinely unique. Traditional methods that depend on population cohorts cannot help these individuals. Evolutionary models can, because they rely on biological principles rather than statistical frequency.
Ranking Pathogenicity Across the Entire Proteome
Early variant effect predictors could determine whether a mutation was likely harmful, but they could not compare severity across different genes. A doctor reviewing a patient’s genome might find ten candidate mutations across ten different genes, with no way to know which one was the primary driver of disease.
Newer models solve this through cross-gene calibration. popEVE combines evolutionary data with human population genetics from resources like gnomAD and the UK Biobank. This produces a unified severity scale that works across all 20,000 human proteins. When tested on more than 31,000 families with children affected by severe developmental disorders, popEVE correctly ranked the known causal mutation as the most damaging variant in 98% of cases . It outperformed AlphaMissense from Google DeepMind in this benchmark.
Integrating Phenotype Prediction with Genotype Analysis
Knowing that a mutation is harmful is not the same as knowing what disease it causes. A new tool called V2P from the Icahn School of Medicine at Mount Sinai addresses this limitation. The model predicts not only pathogenicity but also the likely phenotypic outcome, whether a mutation tends to cause neurological disorders, cancers, or other disease categories.
This capability accelerates diagnostic workflows. Clinicians can filter candidate variants by their predicted phenotypic match to the patient’s symptoms. In tests using real patient data, V2P ranked the true disease-causing variant among the top ten candidates with high consistency. The model operates on a broad categorical level for now, but refinement toward specific disease prediction is underway.
Addressing Non-Coding Mutations Beyond Protein-Coding Regions
Protein-coding DNA accounts for only about 2% of the human genome. The remaining 98% is non-coding, yet mutations in these regions can profoundly disrupt gene expression. Standard clinical sequencing often overlooks these variants because their functional impact is difficult to interpret.
New AI tools are closing this gap. Illumina’s PromoterAI interprets mutations in promoter regions, the non-coding sequences that control where and when genes are transcribed. Combined with whole-genome sequencing, PromoterAI can identify regulatory variants that explain disease in patients whose protein-coding regions appear normal. Similarly, Google DeepMind’s AlphaGenome analyzes non-coding DNA mutations and predicts how they affect nearby gene expression across different tissue types. This tissue-specific capability is crucial because a mutation might disrupt gene function in heart cells but not in brain cells, matching the observed pattern of symptoms.
Clinical Validation and Real-World Diagnostic Yield
Benchmark performance matters, but clinical utility is the ultimate test. Multiple AI models have now been validated on large patient cohorts with demonstrated diagnostic improvements.
When popEVE was applied to approximately 30,000 undiagnosed patients with severe developmental disorders, it provided a diagnosis in about one-third of cases. More significantly, it identified 123 genes never previously linked to developmental disorders. Of these, 104 were observed in only one or two patients worldwide. Twenty-five of these candidate genes have since been independently confirmed by other research laboratories.
The aiDIVA system, developed by researchers in Germany, takes a different approach. It combines random forest classifiers, evidence-based scoring, and large language models to prioritize causal variants. In a benchmark of more than 3,000 solved rare disease cases, aiDIVA-meta placed the correct causal variant in the top three candidates for 97% of cases. The system also provides interpretable explanations, addressing the black-box concern that limits clinician trust in some AI tools.
Addressing Ancestry Bias in Genetic Databases
A persistent problem in genomic medicine is the overrepresentation of European ancestry in reference databases. Many variant prediction tools inadvertently penalize individuals from underrepresented populations, flagging benign population-specific variants as pathogenic simply because they have not been seen before .
popEVE addresses this through its design. By treating all human variants equally regardless of whether they appear once in a specific population or a thousand times in European populations, it produces fewer false positives for underrepresented groups . The model asks whether a mutation has been seen in any human, not whether it is common in European reference data. This approach has significant implications for equitable rare disease diagnosis globally.
What makes this especially valuable is that popEVE can work with only the patient’s genetic information. It does not require parental DNA or large family cohorts . For healthcare systems with limited resources, this makes advanced genetic diagnosis faster, simpler, and more affordable.
Practical Considerations for Clinical Implementation
No AI model replaces clinical judgment. These tools are decision support systems, not autonomous diagnosticians. Each has specific strengths and limitations.
popEVE only interprets missense mutations that alter a single amino acid in a protein. It does not analyze other variant types . aiDIVA requires phenotypic data encoded as Human Phenotype Ontology terms to perform its ranking . PromoterAI and AlphaGenome focus on non-coding regions but require whole-genome sequencing data, which remains more expensive than exome sequencing.
Clinicians considering implementation should ask: Which variant classes are most relevant to my patient population? Do we have the required data types? How will we validate model outputs against clinical findings?
The evidence base supports using these tools as part of a structured diagnostic pipeline, not as standalone answers.
AI Identifying Unknown Genetic Mutations Causing Rare Diseases
Conclusion
The role of AI in rare disease genetics has shifted from research novelty to clinical necessity. With half of all rare disease patients remaining undiagnosed, traditional methods have reached their limits. Evolutionary models, phenotype-integrated systems, and non-coding analyzers are now filling critical gaps. The most useful tools do not simply classify variants as harmful or benign. They rank severity across genes, predict specific disease outcomes, and function equitably across ancestral backgrounds. For clinicians and researchers, the question is no longer whether to use AI for genetic diagnosis, but which combination of tools best serves their patient population. The path forward requires integrating these models into diagnostic workflows while maintaining rigorous clinical validation and human oversight.
FAQs
How accurate are AI models at identifying unknown disease-causing mutations?
In validation studies, leading models like popEVE correctly rank the true causal variant as most damaging in 98% of cases, while aiDIVA identifies the correct variant in the top three candidates for 97% of cases.
Can AI detect mutations that cause diseases never seen before in medical literature?
Yes. Evolutionary models assess mutations based on protein conservation across species rather than prior human cases, enabling detection of genuinely novel pathogenic variants.
Do these AI tools work equally well for all ancestral backgrounds?
Models designed with population-aware calibration, such as popEVE, show reduced ancestry bias by treating all human variants equally regardless of population frequency.


{kind=link}